Vistara test

The objective of the Vistara test is to obtain information on 30 gene mutations that may cause 25 different genetic diseases, which often go undetected during an ultrasound scan and manifest only in the later stages of pregnancy or during the first years of a child’s life.
- What will the Vistara test tell me?
- How reliable is the Vistara test?
- What are the alternatives to the Vistara test?
- When will I receive my test results?
- What results can I expect from the Vistara test?
- Who can be tested and when?
- Who can particularly benefit from the Vistara test?
- What monogenic disorders could be detected by the Vistara test?
How does the Vistara test work?
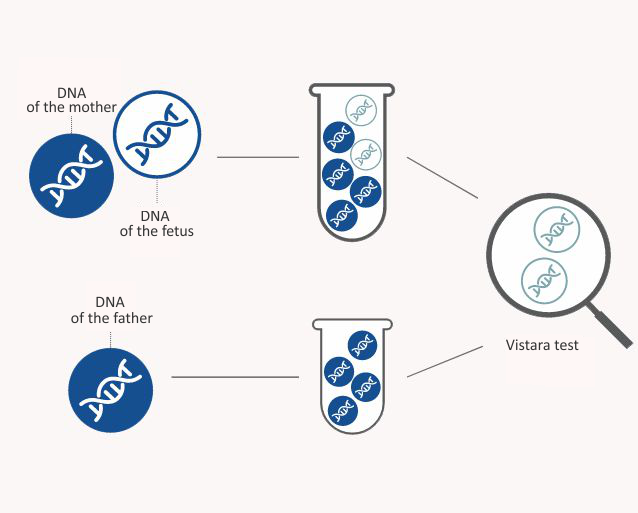
The Vistara test is performed on a mother's blood sample containing both maternal and fetal DNA (genetic material). The fetal DNA to be tested comes from the placenta. The Vistara test detects fetal DNA from the mother’s blood sample and determines whether the fetus has a certain genetic disease or not. This test cannot be used to determine the gender of the fetus.
The Vistara test is a prenatal screening that is completely safe for you and your child. The test is carried out by drawing blood from you, which shall be sent to the Natera laboratory in San Carlos, California by express delivery. A biological father's blood test is not required to perform the Vistara test.
What will the Vistara test tell me?
The Vistara test looks for monogenic disorders, i.e. diseases passed on according to Mendel’s laws of inheritance. Monogenic disorders are caused by a mutation in one or both genes (allele) of a gene pair.
Generally, such diseases are only caused by a gene mutation and the environment plays no role in the occurrence thereof. Such inheritable diseases are also called Mendelian diseases as they are consistent with the laws of inheritance discovered by the monk Gregor Mendel. These diseases are almost exclusively rare, which means that they occur in less than one fetus out of two thousand. Since the Vistara test detects 25 monogenic disorders, the combined occurrence rate of such diseases is one fetus out of six hundred (1: 600). It exceeds the incidence of Down syndrome.
The occurrence of monogenic disorders is based on either the principle of dominant (prevailing) or recessive (hidden) inheritance. If the gene causing the disease is in the autosome (chromosomes 1−22), it is considered autosomal inheritance and the disease occurs in a similar manner regardless of gender.
The Vistara test searches for new mutations (de novo mutations). In this case, the disease gene has not been inherited by either of the parents but has emerged during the formation of the sperm cell or the egg cell. New mutations are especially prominent in autosomal dominant diseases as these are apparent right away – healthy parents surprisingly give birth to an ill child who will, in turn, pass the disease on to their future children in 50% of cases.
The Vistara test is used to check the occurrence of monogenic disorders.
How reliable is the Vistara test?
The accuracy of this screening test in detecting the 30 gene mutations tested is > 99%. The Vistara test is currently the only screening test in Estonia that can be used to detect monogenic disorders in fetuses.
What are the alternatives to the Vistara test?
The Vistara test is a screening for the detection of monogenic disorders. It is not possible to detect fetal chromosome diseases, microdeletions or gender with the Vistara test. The Vistara test is also unable to assess fetal anatomical structures or the complications of pregnancy, such as pre-eclampsia. If you also want to obtain information about fetal chromosome pathologies, microdeletions, fetal organ development, and pregnancy complications, you can combine the Vistara test with the PanoramaXP test and the OSCAR test. For women whose ultrasound scan raises doubts regarding a fetal developmental disorder, invasive diagnostic tests such as chorionic biopsy or amniotic fluid test, which detect > 99% of all chromosomal abnormalities, including rare chromosomal abnormalities that cannot be detected by the Vistara test or other screening tests. In the event of diagnostic studies, it should only be taken into account that in 0.1% of cases, such tests can cause miscarriage, regardless of whether or not the fetus was chromosomal.
When will I receive my test results?
You will receive the test results 21 days after providing your blood sample.
What results can I expect from the Vistara test?
The Vistara test result provided by the Natera laboratory shall contain one of the following statements.
A negative screening result indicates that no pathogenic or likely pathogenic mutations were discovered in the 30 genes tested in your fetus.
- SCREENING POSITIVE
A positive screening result indicates that the likelihood of your fetus having one of the monogenic disorders included in our list is very high, although it is not 100% certain. In the event of such a result, it is necessary to consult with a medical genetic to confirm or exclude the presence of a genetic disease, a diagnostic test such as a chorionic biopsy or an amniotic fluid study or a diagnostic study of the neonatal venous blood at birth is necessary.
Who can be tested and when?
The Vistara test is intended for women of all ages and ethnic backgrounds whose pregnancy has lasted at least 9 full weeks (9 weeks + 0 days).
Vistara test cannot be offered:
- if the mother suffers from one of the gene pathologies screened by the Vistara test;
- in case of twin pregnancy;
- in case of twin pregnancy, if one of the fetuses has perished;
- if the mother has had a blood transfusion in the last month.
The Vistara test can be performed if the biological father of the fetus has been diagnosed with a gene pathology screened by the Vistara test. The Vistara test can also be performed if an ovum or sperm donor was used for fertilization.
Who can particularly benefit from the Vistara test?
The incidence of monogenic disorders does not depend on the woman’s age, however, such diseases are more common when the biological father of the fetus is older.
The test is particularly beneficial to women:
- if the biological father of their child is over the age of 40;
- whose OSCAR test results reveal a higher risk for Down, Edwards or Patau syndrome and for whom a routine chorionic biopsy or amniotic fluid test is indicated for karyotype analysis or submicroscopic analysis;
- if the woman wishes to receive as much information about their child as possible;
- if the fetus exhibits very short long bones or if the cranium of the fetus is very small or is of a peculiar shape (upon suspicion of premature fontanelle closure).
The majority of women who have undergone the Vistara test find out that the risk of their child having a monogenic disorder is very low – this can be very reassuring.
What monogenic disorders could be detected by the Vistara test?
-
Craniosynostosis syndromes
Craniosynostosis is caused by premature ossification of cranial sutures which results in the development of distinct head shape and facial features. In some cases, craniosynostosis may also cause various neurological defects and eating difficulties. The disorder may also increase intracranial pressure and thus lead to impaired vision. Craniosynostosis may be non-syndromic or a part of a genetic syndrome. Approximately 85% of all craniosynostosis cases are non-syndromic. The estimated incidence of craniosynostosis is 1 per 2,000–2,500 live births.
- Antley-Bixler syndrome (FGFR2).
Premature closure of cranial sutures, bowed long bones and fingers, finger joint stiffness. May be accompanied by heart defects, as well as choanal, anal and vaginal atresia. - Apert syndrome (FGFR2).
Premature closure of cranial sutures, finger fusion and dental pathologies occur. - Crouzon syndrome (FGFR2, FGFR3).
Premature closure of cranial sutures, causes hearing loss and dental problems in some cases. - Jackson Weiss syndrome (FGFR2).
Premature closure of cranial sutures and abnormal feet development. - Pfeiffer syndrome, type 1, 2, 3 (FGFR2).
Premature closure of cranial sutures, may be accompanied by hearing loss, mental disability, abnormal development of hands and feet. In the most severe cases, infants may die after birth.
- Antley-Bixler syndrome (FGFR2).
-
Neurological conditions
In the case of neurological conditions, issues occur in the development of the central nervous system which leads to psychomotor retardation and often also intellectual and mental disability in children.
- Epileptic encephalopathy, early infantile, 2 (CDKL5).
Characterized by seizures that start during the first month of life, psychomotor retardation, varying degrees of mental disability, delayed speech development. - Rett syndrome (MECP2).
Occurs primarily in girls. Rapid degeneration of speech and motor skills between 6 to 18 months of age. Autism and epilepsy syndrome are common. - Intellectual disability (SYNGAP1).
Intellectual disability and psychomotor retardation occur.
- Epileptic encephalopathy, early infantile, 2 (CDKL5).
-
Noonan spectrum disorders
Noonan syndrome is an autosomal dominant disorder which is characterized by short stature, congenital heart defects and various degrees of mental retardation. Patients with Noonan syndrome may also have distinct features: short neck, webbed neck, low-set ears, and hypertelorism. There is also a chance of lymphatic dysplasia which causes cystic hygroma and increased nuchal translucency in fetuses. The incidence of Noonan syndrome is approximately 1:1,000 – 1:2,500.
- Cardiofaciocutaneous syndrome (BRAF, MAP2K1, MAP2K2).
Characterized by short stature, distinctive facial features, full body ichthyosis, heart defects. May cause developmental retardation and intellectual disability. - Noonan syndrome 1, 3, 4, 5, 6, 8 (PTPN11, SOS1, RAF1, RIT1, KRAS, NRAS, SOS2, SHOC2, BRAF, MAP2K1, HRAS, CBL).
Characterized by specific facial features inherent to the disease, short stature, skeletal pathology, heard defects, bleeding disorder, undescended testes. In some cases, mild intellectual disability occurs. - Costello syndrome (HRAS).
Characterized by specific facial features inherent to the disorder, short stature, heart defects, developmental and intellectual disability and increased risk of malignancies. - LEOPARD syndrome 1,2 (PTPN11, RAF1).
Similar to Noonan syndrome with significant brown spots on the skin (lentigines), short stature, heart defects, bleeding, possible intellectual disability with varying degree. - Juvenile myelomonocytic leukemia (PTPN11).
Occurrence of rapidly progressing and aggressive leukemia. Five year survival rate 50%.
- Cardiofaciocutaneous syndrome (BRAF, MAP2K1, MAP2K2).
-
Skeletal disorders
There are over 200 congenital bone growth disorders. The most common skeletal disorders are osteochondrodysplasias which affect long bones, the spine, as well as cartilaginous parts. These disorders are also often accompanied by impaired growth, as well as deformed bones and vertebral column in children. However, the mental development of children is oftentimes age-appropriate. In the most severe cases, children die in utero or shortly after birth due to respiratory failure.
- Achondroplasia (FGFR3).
A bone growth disorder characterized by dwarfism with especially short humeri and femurs, a disproportionately big head, and possible spinal stenosis. - CATSHL syndrome (FGFR3).
Characterized by tall stature, scoliosis, hearing loss, finger joint stiffness. May be accompanied by intellectual disability. - Osteogenesis imperfecta, type I, II, III, IV (COL1A1, COL1A2).
Extremely brittle bones, often without no apparent reason. In the most severe cases, infants die due to respiratory failure. - Ehlers-Danlos syndrome, classic, type VIIA, cardiac valvular form, type VIIB (COL1A1, COL1A2).
Characterized by specific facial features inherent to the disorder, connective tissue damage, hypermobile joints. May be accompanied by life-threatening complications such as an aortic aneurysm. - Hypochondroplasia (FGFR3).
A bone growth disorder accompanied by short stature and spinal stenosis. May be accompanied by an epilepsy syndrome along with secondary developmental retardation. - Thanatophoric dysplasia, types I, II (FGFR3).
Severe stunting and skeletal pathology. Often die in the womb or shortly after birth due to respiratory failure.
- Achondroplasia (FGFR3).
-
Congenital syndromes
Such syndromes refer to sets of symptoms characteristic of a certain disorder or the combination thereof. Syndromes often affect various organ systems at a time and are linked to mental retardation.
- Alagille syndrome (JAG1).
Heart defects and liver problems occur. May be accompanied by growth issues and malformation of the spine. - CHARGE syndrome (CHD7).
Choanal atresia, retinal pathology, heart defects, genital abnormalities, abnormal ears, hearing loss, growth and developmental retardation and cleft lip and/or palate occur. - Cornelia de Lange syndrome 1, 2, 3, 4, 5 (NIPBL, SMC1A, SMC3, RAD21, HDAC8).
Characterized by specific facial features inherent to the disorder, growth and developmental retardation, varying degrees of intellectual disability. - Muenke syndrome (FGFR3).
Premature closure of cranial sutures, may be accompanied by hearing loss, psychomotor retardation and cleft lip and/or palate. - Sotos syndrome 1 (NSD1).
Rapid growth, psychomotor, cognitive and mental retardation. Disorders falling into the autism spectrum may occur. - Tuberous sclerosis 1, 2 (TSC1, TSC2).
Multisystem disorder that causes hamartoma in the brain, skin, kidneys, lungs. Growing hamartoma in the brain cause epilepsy syndrome, learning difficulties, psychomotor developmental retardation, autism, behavioral problems.
- Alagille syndrome (JAG1).