Vistara test

Vistara test on sünnieelne mitteinvasiivne sõeltest, mille abil on võimalik teada saada loote 30 geenimutatsiooni kohta, mis võivad põhjustada 25 erinevat geenihaigust, mida väga sageli ei ole ultraheliuuringul võimalik avastada ja mis tihti avalduvad alles raseduse hilises perioodis või lapse esimestel eluaastatel.
- Kuidas Vistara test töötab?
- Mida Vistara test mulle ütleb?
- Kui usaldusväärne on Vistara test?
- Mis on Vistara testi alternatiivid?
- Millal ma oma testi tulemused teada saan?
- Milliseid tulemusi ma Vistara testist oodata võin?
- Keda saab testida ja millal?
- Kes saavad Vistara testist eriti kasu?
- Milliseid monogeenseid haigusi võib leida, kui ma teen Vistara testi?
Kuidas Vistara test töötab?
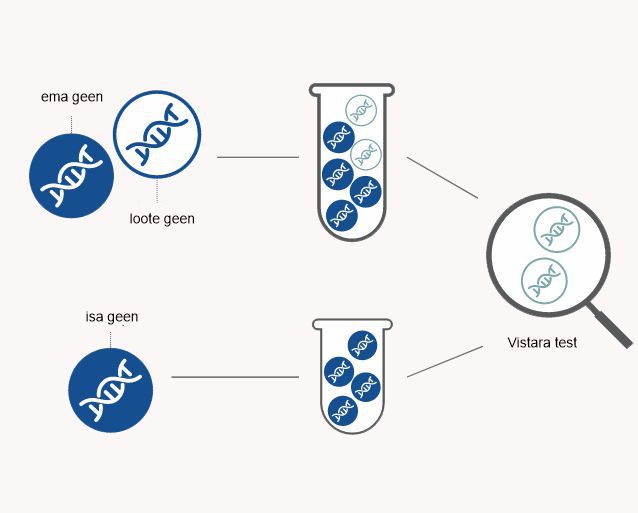
Vistara testi teostatakse ema vereproovist, milles leidub nii ema kui ka loote DNA-d (geneetilist materjali). Loote testitav DNA pärineb platsentast. Vistara test tuvastab ema vereproovist loote DNA ja hindab selle abil, kas lootel on otsitud geenihaigus või mitte. Selle testiga ei saa tuvastada loote sugu. Vistara testi saab kasutada ka siis, kui viljastumisel kasutati munaraku- või spermadoonorit. Bioloogilise isa vereanalüüs pole Vistara testi teostamiseks vajalik.
Vistara test on mitteinvasiivne sünnieelne sõeltest, mis on Sinu ja lapse jaoks täiesti ohutu. Testi tegemiseks võetakse Sinu käsivarrelt vereproov, mis saadetakse kiirkulleriga Californiasse San Carlosesse Natera laborisse.
Mida Vistara test mulle ütleb?
Vistara test otsib monogeenseid ehk Mendeli seaduste järgi päranduvaid haigusi. Monogeenseid haigusi põhjustab geenipaari ühes või mõlemas geenis (alleelis) olev mutatsioon.
Üldiselt on need haigused ainult geenimutatsiooni põhjustatud ja keskkonnal ei ole nende tekkel tähtsust. Sel viisil päranduvaid haigusi nimetatakse ka Mendeli haigusteks, sest need järgivad munk Gregor Mendeli avastatud pärilikkuse seaduspärasusi. Need kuuluvad peaaegu eranditult harvaesinevate haiguste hulka, mis tähendab, et neid esineb harvemini kui ühel lootel kahest tuhandest. Kuna Vistara test otsib 25 monogeenset haigust, on haiguste kombineeritud esinemissagedus üks loode kuuesajast (1 : 600). See on suurem kui Downi sündroomi esinemissagedus.
Monogeensete haiguste avaldumine järgib kas dominantse (valitseva) või retsessiivse (varjatud) pärilikkuse põhimõtet. Kui haigust põhjustav geen paikneb autosoomis (kromosoomid 1−22), on tegu autosoomse pärilikkusega ja haigus avaldub ühte moodi soost sõltumata.
Vistara test otsib uusmutatsioone (de novo-mutatsioonid). Nende puhul pole haigusgeen päritud kummaltki vanemalt, vaid on tekkinud seemneraku või munaraku moodustumise käigus. Uusmutatsioonid on eriti silmatorkavad just autosoomsete dominantsete haiguste korral, sest tulevad esile kohe – üllatuslikult saavad terved vanemad haige lapse, kes teoreetiliselt annab 50% juhtudel haiguse omakorda edasi enda tulevastele lastele.
Kui usaldusväärne on Vistara test?
See sõeltest suudab tuvastada üle 99% testiga uuritavast 30 geenimutatsioonist. Vistara test on sõeltest, mis on suuteline lootel tuvastama monogeenseid haigusi.
Mis on Vistara testi alternatiivid?
Vistara test on sõeltest monogeensete haiguste avastamiseks. Vistara testiga pole võimalik tuvastada loote kromosoomihaigusi, mikrodeletsioone ega sugu. Vistara testiga pole võimalik hinnata ka loote anatoomilisi struktuure ega rasedusega kaasneda võivaid tüsistusi nagu preeklampsia ja loote kasvupeetus. Kui Sa soovid teavet ka loote kromosoomi patoloogiate, mikrodeletsioonide, loote organstruktuuride arengu ja rasedusaegsete komplikatsioonide kohta, on võimalik Vistara testi kombineerida PanoramaXP testi ja OSCAR-i testiga. Naiste puhul, kelle ultraheliuuringus jääb kahtlus loote arengurikke suhtes, on kasutusel sellised invasiivsed diagnostilised testid nagu koorionibiopsia või looteveeuuring, mis tuvastavad üle 99% kõigist kromosoomianomaaliatest, sealhulgas ka haruldasi kromosoomianomaaliaid, mida Vistara testiga ega ka teiste sõeltestidega avastada ei saa. Diagnostiliste uuringute korral tuleb ainult arvestada sellega, et 0,1%-l juhtudest võivad need põhjustada raseduse katkemise sõltumata sellest, kas loode oli kromosomaalse patoloogiaga või mitte.
Millal ma oma testi tulemused teada saan?
Testi tulemused saad Sa teada 21 päeva möödudes pärast vereanalüüsi andmist.
Milliseid tulemusi ma Vistara testist oodata võin?
Natera laborist saadav Vistara testi vastus sisaldab ühte järgmistest võimalustest.
Sõeluuringu negatiivne tulemus tähendab, et Sinu lootel ei tuvastatud Vistara testiga uuritavas 30 geenis patogeenset või tõenäoliselt patogeenset mutatsiooni.
- SÕELUURING POSITIIVNE
Sõeluuringu positiivne tulemus tähendab, et Sinu lootel on ühe Vistara testiga avastatava monogeense haiguse esinemise tõenäosus väga kõrge, aga see pole 100% kindel. Sellise vastuse korral on kinnitada või välistada geenihaiguse olemasolu, tuleb teha diagnostiline test, näiteks koorionbiopsia või looteveeuuring või sünnijärgselt vastsündinu veeniverest teostatav diagnostiline uuring.
Keda saab testida ja millal?
Vistara test on mõeldud igas vanuses ja igasuguse etnilise päritoluga naistele, kelle rasedus on kestnud vähemalt 9 täisnädalat (9 nädalat + 0 päeva).
Vistara testi ei saa pakkuda:
- kui ema põeb ühte Vistara testiga uuritavat geenipatoloogiast;
- kaksikraseduse korral;
- kaksikraseduse korral, kui üks loode on hukkunud;
- kui emale on viimase kuu jooksul tehtud vereülekanne.
Vistara testi saab teha, kui loote bioloogilisel isal on diagnoositud Vistara testiga uuritav geenipatoloogia. Samuti saab teostada Vistara testi ka siis, kui viljastumisel kasutati munaraku- või spermadoonorit.
Kes saavad Vistara testist eriti kasu?
Monogeensete haiguste esinemissagedus ei sõltu naise vanusest, küll aga esinevad need haigused sagedamini loote vanemaealise bioloogilise isa puhul. Soovitame Vistara testi teostamist, kui lapse isa vanus on üle 40 aasta.
- kelle lapse bioloogiline isa on vanem kui 40 aastat;
- kellel OSCAR-i testis on kõrgenenud risk Downi, Edwardsi või Patau sündroomiks ning kellel on näidustatud diagnoosi täpsustamiseks rutiinne koorionibiopsia või looteveeuuring karüotüübi hindamiseks või submikroskopiaalseks analüüsiks;
- kui naine soovib oma lapse kohta saada nii palju informatsiooni kui võimalik;
- kui kolmanda trimestri ultraheliuuringul on lootel väga lühikesed toruluud või kui loote ajukolju on väga väike või esineb selle väga ebatavaline kuju (kui on kahtlus koljulõgemete enneaegsele sulgumisele).
Enamik naistest, kellele Vistara test on tehtud, saavad teada, et nende lapsel on testitud monogeense haiguse tekkerisk väike – mis võib olla väga julgustav.
Milliseid monogeenseid haigusi võib leida, kui ma teen Vistara testi?
-
Kraniosünostoosi sündroomid
Kraniosünostoosi põhjustab koljuluu õmbluste enneaegne luustumine, mille tulemusena arenevad iseloomulik peakuju ja näojooned. Mõningatel juhtudel võib kraniosünostoos põhjustada neuroloogilisi häireid ja söömisraskusi. Haigus võib suurendada ka koljusisest rõhku ning põhjustada nägemishäireid. Kraniosünostoos võib esineda mittesündroomsena või ka osana geneetilistest sündroomidest. Ligikaudu 85% kõigist kraniosünostoosi juhtumitest on mittesündroomsed. Kraniosünostoosi esineb hinnanguliselt 1 : 2000-2500 elussünni kohta.
- Antley-Bixleri sündroom (FGFR2).
Esineb koljuluude enneaegne luustumine, kõverad toruluud ja sõrmed, sõrmeliigeste jäigastumine. Võib kaasneda südame arengurikkeid, pärakuavause, ninasõõrmete ja tupe puudumine. - Aperti sündroom (FGFR2).
Esineb koljuluude enneaegne luustumine ja sõrmede omavaheline kokkukasvamine ja hambumuse patoloogia. - Crouzoni sündroom (FGFR2, FGFR3).
Esineb koljuluude enneaegne luustumine, põhjustab mõnedel juhtudel kuulmiskadu ja hambumisprobleeme. - Jackson-Weissi sündroom (FGFR2).
Esineb koljuluude enneaegne luustumine ja kõrvalekalded jalgade arenemises. - Pfeifferi sündroom, tüüp 1, 2, 3 (FGFR2).
Esineb koljuluude enneaegne luustumine, võib esineda kuulmiskadu, vaimset puuet, kõrvalekalded käte ja jalgade arenemises. Raskematel juhtudel võib vastsündinu sünnijärgselt hukkuda.
- Antley-Bixleri sündroom (FGFR2).
-
Neuroloogilised seisundid
Neuroloogiliste seisundite puhul esinevad probleemid kesknärvisüsteemi arengus, mille tagajärjel ilmneb lastel psühhomotoorne arengupeetus ja sageli vaimne puue.
- Epileptiline entsefalopaatia, varajane infantiilne, 2 (CDKL5).
Iseloomulikud on esimesel elukuul algavad krambihood, psühhomotoorne arengupeetus, erineva raskusastmega vaimne alaareng, aeglane kõne areng. - Retti sündroom (MECP2).
Enamusjuhtudel esineb tüdrukutel. Esineb rääkimise ja motoorsete oskuste kiire taandareng 6 kuni 18 kuu vanuses. Sageli esineb autism ja krambisündroom. - Vaimne puue (SYNGAP1).
Esineb vaimne puue ja psühhomotoorne arengupeetus
- Epileptiline entsefalopaatia, varajane infantiilne, 2 (CDKL5).
-
Noonani spektri häired
Noonani sündroom on autosoom-dominantselt päranduv haigus, mida iseloomustab lühike kasv, kaasasündinud südamerike ning erinevas raskusastmes vaimse arengu peetus. Noonani sündroomiga patsientidel on ka iseloomulik välimus: lühike kael, nahavolt kaelal, madala asetsusega kõrvad, hüpertelorism. Lisaks võib esineda lümfisüsteemi düsplaasia, mis on aluseks tsüstilise hügroomi ja kuklavoldi suurenemisele lootel. Noonani sündroomi esinemissagedus on ~1 : 1000-1 : 2500.
- Kardio-fatsio-kutaanne sündroom (BRAF, MAP2K1, MAP2K2).
Iseloomulikud on lühike kasv, muutunud näojooned, kogukeha ihtüoos, südame arengurikked. Võib põhjustada arengupeetust ja vaimset puuet. - Noonani sündroom 1, 3, 4, 5, 6, 8 (PTPN11, SOS1, RAF1, RIT1, KRAS, NRAS, SOS2, SHOC2, BRAF, MAP2K1, HRAS, CBL).
Iseloomulikud on spetsiifilised antud haigusele tüüpilised näojooned, lühike kasv, skeletipatoloogia, südame arengurikked, veritsushäire, munandite laskumishäire. Mõnedel juhtudel kerge vaimne puue. - Costello sündroom (HRAS).
Iseloomulikud on spetsiifilised antud haigusele tüüpilised näojooned, lühike kasv, südame arengurikked, arengu- ja vaimupuue ning pahaloomuliste kasvajate esinemise võimaluse suurenemine. - LEOPARDI sündroom 1, 2 (PTPN11, RAF1).
Sarnaneb Noonani sündroomile, märkimisväärsete pruunide laikudega nahal (lentiginoos), lühike kasv, südame arengurikked, veritsemine, võimalik erineva raskusastmega vaimne puue. - Juveniilne müelomonotsüütiline leukeemia (PTPN11).
Esineb kiire kuluga, agressiivne leukeemia. 5 aasta elulemus 50%.
- Kardio-fatsio-kutaanne sündroom (BRAF, MAP2K1, MAP2K2).
-
Luustikuga seotud häired
Kaasasündinud luude kasvuhäiretega sündroome on üle kahesaja erineva vormi. Sagedasemateks luustikuga seotud häireteks on osteokondrodüsplaasiad, mis haaravad nii pikki toruluid, selgroogu kui ka luude kõhrelisi komponente. Sageli kaasneb selle haigustega laste kasvupeetus, deformeerunud luud ja lülisammas. Samas laste vaimne areng on sageli eakohane. Raskemate vormide puhul hukkuvad lapsed üsasiseselt või vahetult sünnijärgses perioodis hingamispuudulikkuse tagajärjel.
- Akondroplaasia ehk kõhrevaegmoodustus (FGFR3).
Esineb luustiku kasvuhäire, millele on iseloomulikud väljendunud kääbuskasv, eriti on lühenenud õlavarreluu ja reieluu, pea on ebaproportsionaalselt suur, esineb seljaajukanali ahenemist. - CATSHL-i sündroom (FGFR3).
Iseloomulikud on pikk kasv, selgroo külgkõverdus, kuulmiskadu, sõrmeliigeste jäigastumine. Võib kaasneda vaimne puue. - Osteogenesis imperfecta, tüüp I, II, III, IV (COL1A1, COL1A2).
Äärmiselt kergesti purunevad luud, sageli teadaoleva põhjuseta. Raskematel juhtudel vastsündinud hukkuvad hingamispuudulikkuse tagajärjel. - Ehlers-Danlosi sündroom, klassikaline, tüüp VIIA; südameklapi vorm, tüüp VIIB (COL1A1, COL1A2).
Iseloomulikud on spetsiifilised antud haigusele iseloomulikud näojooned, sidekoe kahjustused, hüpermobiilsed liigesed. Võib kaasneda eluohtlik tüsistus nagu aordi lõhustav aneurüsm. - Hüpokondroplaasia (FGFR3).
Luustiku kasvuhäire, millega kaasneb lühike kasv ja seljaajukanali ahenemine. Võib kaasneda krambisündroom koos sekundaarse arengupeetusega. - Tanatroofiline düsplaasia, tüübid I, II (FGFR3).
Raske kasvupeetus ja skeletipatoloogia. Sageli hukkuvad emaüsas või sünnijärgses perioodis hingamispuudulikkuse tõttu.
- Akondroplaasia ehk kõhrevaegmoodustus (FGFR3).
-
Kaasasündinud sündroomid
Sündroomide all mõeldakse kindale haigusele iseloomulike haigustunnuste kogumikke või nende kombinatsioone. Sageli haaravad sündroomid mitmeid elundsüsteeme korraga ja on seotud vaimse alaarenguga.
- Alagille´i sündroom (JAG1).
Esinevad südame arengurikked ja maksaprobleemid. Võib kaasneda kasvuprobleeme ja lülisamba kõrvalekaldeid. - CHARGE´i sündroom (CHD7).
Esineb ninakäikude sulgus, silma võrkkesta patoloogia, südame arengurikked, suguelundite kõrvalekalded, abnormsed kõrvad, kuulmiskadu, kasvu- ja arengupeerus ning huule- ja/või suulaelõhe. - Cornelia de Lange´i sündroom 1, 2, 3, 4, 5 (NIPBL, SMC1A, SMC3, RAD21, HDAC8).
Iseloomulikud on spetsiifilised antud haigusele tüüpilised näojooned, kasvu- ja arengupeetus, erineva raskusastmega vaimne puue. - Muenke´i sündroom (FGFR3).
Esineb koljuluude enneaegne luustumine, võib esineda kuulmiskadu, psühhomotoorne arengupeetus ning huule- ja/või suulaelõhe. - Sotose sündroom 1 (NSD1).
Kiire kasv, esineb psühhomotoorne, kognitiivne ja vaimne alaareng. Võib esineda autismispektri häireid. - Tuberoosne skleroos 1, 2 (TSC1, TSC2).
Multisüsteemne haigus, mille puhul tekivad hamartoomid ajus, nahas, neerudes, kopsudes. Hamartoomide tekkel ajus esineb krambisündroom, õppimisraskused, psühomotoorne arengupeetus, autism, käitumisprobleemid
- Alagille´i sündroom (JAG1).