Loote ultrahelikeskuses on võimalik avastada DiGeorge´i sündroomiga looteid
22q11.2 mikrodeletsioonisündroom ehk DiGeorge’i sündroom on inimese sagedaseim deletsioonisündroom, mis esineb ligikaudu 1:2000 – 1:4000 elusalt sündinud lapsel ja võib põhjustada mitmesuguseid kaasasündinud arengurikkeid ning ka keskmist kuni mõõdukat vaimupuuet.
DiGeorge´i sündroom on esinemissageduselt teine sagedaseim kromosomaalne patoloogia, mis põhjustab lootel kaasasündinud südame arengurikkeid, jäädes alla vaid Downi sündroomile. Need kaks sündroomi on ka muus osas sarnased, kuna mõlemal on palju haigustunnuseid ja sealhulgas kehv lihastoonus.
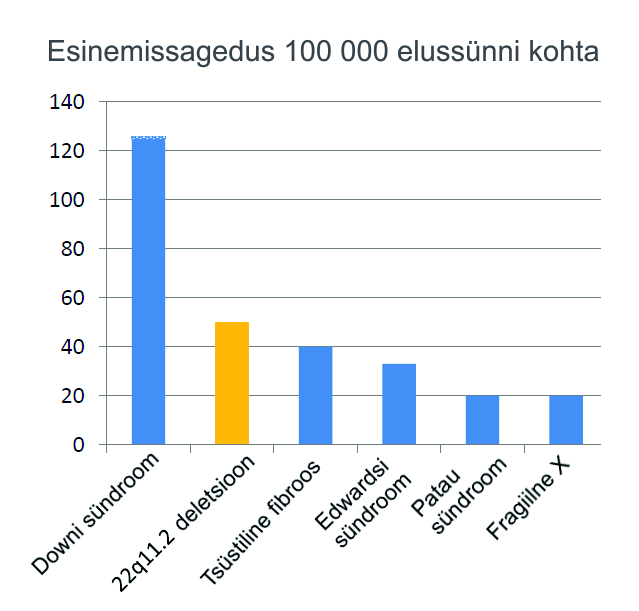
Mis on DiGeorge’i sündroom?
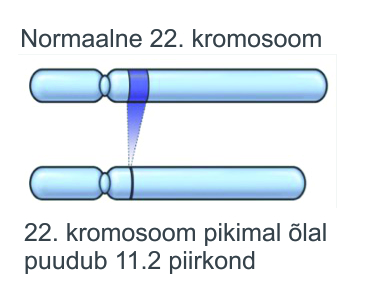
DiGeorge’i sündroomi põhjustab 22. kromosoomi pikema õla väikese osa (lookus 22q11.2) puudumine (mikrodeletsioon).
Sellest tulenevad muutused mõjutavad suuremat osa kehast. Suur osa sündroomi põdevatest lastest omavad südamerikkeid, immuunhäireid ja iseloomulikke, kuigi sageli vaevumärgatavaid näojooni. Peaaegu kõigil sündroomiga lastest on keskmine kuni mõõdukas vaimne puue ning aeglustunud keeleline ja kõneline areng. Mõnedel lastel on kaltsiumipuudus, neeruprobleemid, söömisraskused, esinevad krambid või muud terviseprobleemid. Ligikaudu ühel viiest (20%) DiGeorge’i sündroomiga lapsest on autismispektri häire ning üks neljast (25%) noorest täiskasvanust põeb psühhiaatrilist haigust, näiteks skisofreeniat. Tõsiste südameprobleemide või immuunsüsteemihäiretega imikutel on kõrgem suremisoht. DiGeorge’i sündroomiga inimestel, kes lapseea üle elavad, võib olla lühem eluiga ja kõrgem äkksurma oht.
Mis on 22q11.2 deletsioonisündroomi põhjuseks?
Enamikul DiGeorge’i sündroomiga lastest esineb 3 MB deletsioon (hõlmab enda alla keskmiselt 40 geeni) 22. kromosoomi ühel koopial. 22q11.2 deletsioon toimub juhuslikult ( de novo mutatsioon) ja ei ole enamasti pärilik. Siiski pärisid teadaolevalt ligikaudu 5-10% sündroomiga lastest selle oma ühelt vanemalt, kes põeb sama seisundit ( haiguse ülekandmise võimalus vanemalt lapsele 50%). Mõlema vanema testimine võib aidata määrata sellise olukorra uuesti esinemise tõenäosuse järgmise raseduse korral.
Miks on DiGeorge’i sündroom´i diagnoosimine oluline?
Järgnevalt loetletud seisundite ravimine ja mõningatel juhtudel füsioteraapia, tööteraapia ning eriõppe kombineerimine võib 22q11.2 deletsioonisündroomiga patsientide elukvaliteeti tõsta.
- Hüpokaltseemia. Madal kaltsiumi tase ehk hüpokaltseemia on DiGeorge’i sündroomi puhul levinud, eelkõige just vastsündinutel. See on tingitud kõrvalkilpnäärmete alaarengust tingitud puuduliku talitusega. Paratüroidhormooni puudulikkusest tingitud kaltsiumipuudus võib põhjustada krampe. Sageli jäetakse madalast kaltsiumitasemest tingitud krampide põhjused tähelepanuta, mistõttu probleem võib jääda ravita ja mõjutada lapse vaimset arengut. DiGeorge’i sündroomiga vastsündinutel tuleb jälgida hüpokaltseemia märke ja kui need tuvastatakse, tuleb lapsele koheselt ravi määrata. Hüpokaltseemia võib kasvuspurtide, puberteedi või haiguste ajal ning operatsioonide järgselt korduda.
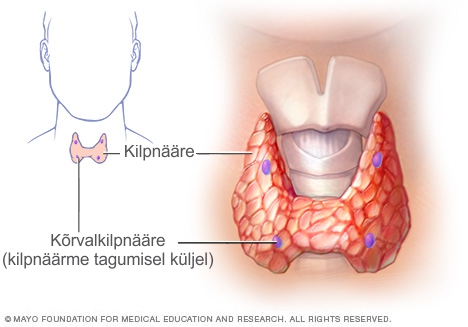
- Immuunpuudulikkus.
Lümfaatiline süsteem on immuunsüsteemi oluline osa, mis kaitseb last infektsioonide eest. Lümfaatilise süsteemi osadeks on põrn, tüümus (harknääre), lümfisõlmed ja lümfiteed ning samuti tonsillid ja adenoidid. DiGeorge’i sündroomiga lastest on ligi 75% tüümuse alaareng ja sellega tingitud T- rakuline immuunpuudulikkus. Immuunpuudulikkuse ohu tõttu tuleks sündroomi põdevaid lapsi enne elusviirusvaktsiinide manustamist kontrollida.
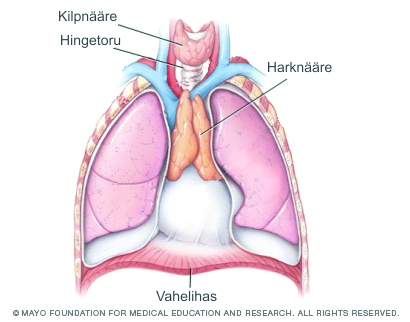
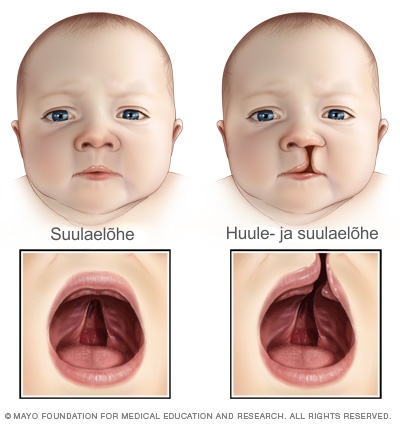
- Suulae väärarengud.
Ligi 75% sündroomi põdevatest lastest on suulae probleemidega, mis võib olla kas struktuurne või funktsionaalne või nende kombinatsioon. Selline probleem võib põhjustada söömisraskusi ja/või kõnehäireid. Kui probleemi varakult ei korrigeerita, võib see mõjutada kõne arengut. Enamasti on selline seisund ravitav.
- Söömisraskused. DiGeorge’i sündroomiga vastsündinutel on sageli söömisraskused, mis ei ole tingitud suulaest ega südamehäiretest. Söömisraskuse põhjuseks võib olla kõri- ja söögitoru lihaste motoorikahäire, mille levinumateks sümptomiteks on refluks ning kõhukinnisus. Harva võivad esineda soolte malrotatsioon ja Hirschsprungi tõbi. Enamik nendest probleemidest on ravitavad.
- Kaasasündinud südamerikked. Ligikaudu 75% sündroomiga isikutest on kaasasündinud südamerikkega, mis ongi sageli antud sündroomi kahtlustamise aluseks. Kui on diagnoositud DiGeorge’i sündroom, tuleb lootel teostada südame ehhokardiograafia südame arengurikete olemasolu/raskusastme hindamiseks. Kui lootel avastatakse südamerike, suunatakse ta lastekardioloogi konsultatsioonile edasiste jälgimis- ja raviplaanide koostamiseks. Paljud südamerikked on tänapäeval opereeritavad.
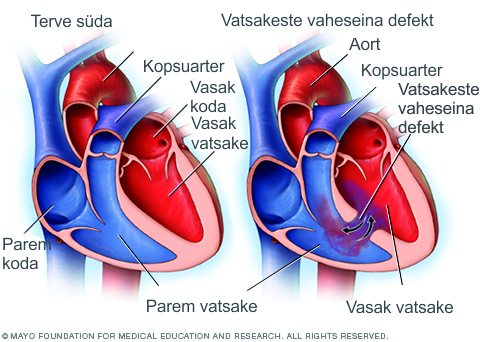
- DiGeorge’i sündroomiga kaasnevad ka muud meditsiinilised probleemid, sealhulgas mõlemapoolne neeruvaagnate laienemine, kuulmiskadu, kõrva-nina-kurgu komplikatsioonid, autoimmuunhaigused, kasvupeetus ning skeletiprobleemid, millega kaasnevad selgroo- ja jäsemete patoloogiad.
Kuidas peaksin suhtuma Panorama testi tulemusse kui see näitab suurenenud riski DiGeorge’i sündroomi olemasolusse minu lootel ?
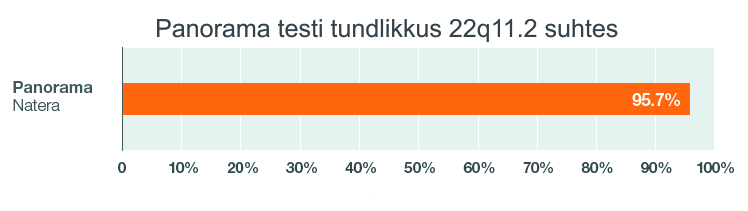
Panorama testi tundlikkus 22q11.2 mikrodeletsiooni avastamisel on 95,7%. ja spetsiifilisus > 99%. Seega tegemist on täpse skriiningtestiga.
Kuid vaatamata oma täpsusele jääb Panorama test ikkagi skriiningtestiks. Ultrahelimarkerite puudumisel on Panorama testi positiivne ennustav väärtus kõigest 20%. Samas positiivse ultrahelileiu puhul (südamerike, alaarenenud väike harknääre) on Panorama testi positiivne ennustav väärtus ligilähedane 100%-le. Seega tegemist pole diagnostilise testiga. Kõrge riskiga Panorama testi korral peab teostama diagnoosi kinnitava looteveeuuringu või koorionibiopsia. Submikroskoopilise kromosoomianalüüsi alusel, on võimalik avastada 22q11.2 deletsioonisündroomi põhjustavat 22. kromosoomi puuduvat osa. Juhul, kui last ootav naine ei soovi rasedusaegset invasiivset uuringut, võib ka sünnijärgselt vastsündinu veeniverest saadud kromosoome uurida submikroskoopilise kromosoomianalüüsi abil.
Panorama testi negatiivne ennustav väärtus 22q11.2 suhtes on 99,9%. Seega võimalus, et Panorama test haigust ära ei tunne, on väga väike.
Panorama testi tundlikkus DiGeorge sündroomi avastamisel sõltub loote rakuvälise DNA hulgast. Kui loote rakuvälise DNA hulk skriiningnegatiivsetel naistel on suurem kui 6,5%, on lootel 22q11.2 mikrodeletsiooni testi järgne risk 1: 9000 sünnituse kohta, mida võib lugeda väga rahustavaks.
Kas DiGeorge’i sündroomi saab testida ka kaksikraseduse puhul?
DiGeorge’i sündroomi saab testida ainult monokoriaalsete kaksikute puhul (kaksikud, kes jagavad ühte platsentat ja on alati ühemunaraku kaksikud). DiGeorge’i sündroomi ei saa testida dikoriaalsete kaksikute puhul (kaksikud, kellel mõlemal on oma platsenta ning võivad olla nii ühemunaraku- kui ka erimunaraku kaksikud) ja IVF raseduse puhul, kui on kasutatud doonormunarakke.